本期导读代谢研究热点
胆汁酸调节免疫:24-去甲熊去氧胆酸NorUDCA化身“炎症克星”,剑指TH17细胞并逼其改邪归正;AKR1D1则暗中改造iso-LCA,激活NK细胞上演肝癌剿灭战;
二甲双胍降糖抗抑郁:降糖机制新解——充当“葡萄糖流量调度员”将血糖赶入肠道;胍胍还可以影响肠菌作用于胆汁酸—色氨酸代谢缓解动物抑郁;
神经酰胺代谢调节受体:代谢功能研究主刊频出,它一边通过FPR2受体抑制脂肪细胞产热,同时另一边它被血管受体识别从而引发动脉硬化危机;
抗衰明星氨基酸:瓜氨酸调节代谢助力巨噬细胞的对抗衰老炎症;另一种来自蘑菇的氨基酸——麦角硫因给线粒体装上“涡轮增压”,让小鼠秒变运动达人。
导读目录
1.GUT丨24-去甲熊去氧胆酸通过靶向TH17致病性和转分化改善肠道炎症
2.Cell Metabolism丨AKR1D1通过促进胆汁酸代谢介导的NK细胞毒性抑制肝癌进展
3.Communications Medicine | 二甲双胍调节从血液循环到肠腔的葡萄糖通量
4.Brain Behavior and Immunity | 二甲双胍通过肠道微生物衍生的胆酸代谢物重编程色氨酸代谢,以改善小鼠的抑郁样行为
5.Science | 神经酰胺通过FPR2受体的代谢信号传导抑制脂肪细胞产热
6.Nature | 内源性受体CYSLTR2和P2RY6感知神经酰胺来加重动脉粥样硬化
7.Science Advances | 瓜氨酸调节小鼠巨噬细胞代谢和炎症以对抗衰老
8.Cell Metabolism | 麦角硫因通过激活MPST调控线粒体功能和运动表现
资源领取
本期导读文献原文,请在公众号后台回复“2025年3月绘谱导读”,即可获取资源链接。
【1】
GUT丨24-去甲熊去氧胆酸通过靶向TH17致病性和转分化改善肠道炎症

原发性硬化性胆管炎(PSC)是一种免疫介导的胆汁淤积性肝病,常伴随肠道炎症,但目前缺乏有效治疗手段。T辅助17型(T_H17)细胞驱动的炎症反应被认为是PSC及其相关肠道疾病的关键病理机制。本研究聚焦新型胆汁酸24-去甲熊去氧胆酸(NorUDCA),发现其通过抑制T_H17细胞致病性、促进调节性T细胞(Tregs)生成,显著缓解肠道炎症,为PSC及相关免疫疾病的治疗提供了新方向。
1. 首先通过CD4+初始T细胞(T_Naive)过继转移模型评估NorUDCA对T_H17分化的影响。发现NorUDCA干预显著减少肠道T_H17细胞比例,同时增加Tregs丰度。组织学分析显示其可减轻肠道验证、减少杯状细胞丢失、改善黏膜屏障;此外,次级淋巴器官中T_H17/Tregs比例也同步改变。
2. 对αCD3刺激模型补充NorUDCA,发现肠道促炎性细胞的生成减少,而 T_H17向Tregs和Tr1细胞的转化增多。机制研究表明,NorUDCA抑制mTORC1信号通路,减少核糖体蛋白S6(RPS6)磷酸化,从而限制T_H17的代谢活性。DEREG小鼠实验证实:Tregs缺失后,NorUDCA对肠道炎症和体重减轻的保护作用显著减弱,证明其疗效依赖于Tregs的生成。
3. 体外实验进一步揭示NorUDCA抑制CD4+T_Naive细胞向IL-17A+细胞分化,并诱导Foxp3+Tregs。代谢组学和转录组学分析显示,NorUDCA通过抑制谷氨酰胺代谢-mTORC1-糖酵解轴,降低α-KG水平,从而抑制T_H17的增殖和效应功能。此外还发现添加α-KG可逆转NorUDCA对mTORC1活性和Tregs生成的调控作用,证实代谢重塑是其核心机制。
4. 最后构建人源化NSG小鼠模型验证NorUDCA的临床转化潜力。结果显示,NorUDCA治疗显著减少肠道和中枢神经系统中T_H17细胞浸润,并增加Tregs比例;一些关键代谢指标,如RPS6磷酸化和Ki67表达受到抑制,提示其具有广泛适用性。

参考文献:Zhu C, Boucheron N, Al-Rubaye O, et al. 24-Nor-ursodeoxycholic acid improves intestinal inflammation by targeting TH17 pathogenicity and transdifferentiation. Gut. 2025
【2】
Cell Metabolism丨AKR1D1通过促进胆汁酸代谢介导的NK细胞毒性抑制肝癌进展

肝细胞癌(HCC)的发生发展与胆汁酸代谢紊乱和免疫抑制微环境密切相关,但二者间的调控机制尚未明确。本研究通过多组学分析和动物模型实验,揭示了胆汁酸代谢和肠道菌群平衡可维持NK(自然杀伤)细胞的抗肿瘤功能,并提出靶向异石胆酸(iso-LCA)及其来源菌B.ovatus可增强HCC免疫治疗效果。
1. 首先对33例HCC患者配对样本进行WB和免疫组化分析,发现肿瘤组织中AKR1D1(醛酮还原酶1D1,主要参与类固醇激素的代谢)表达水平显著降低;随后发现敲除AKR1D1后荷瘤小鼠自发肝癌发生率显著升高;TCGA数据库分析进一步表明AKR1D1低表达的HCC患者生存期更短。表明AKR1D1缺失会促进HCC的进展。
2. 随后对AKR1D1敲除小鼠的肝脏组织进行转录组测序,发现NK细胞介导的细胞毒性相关通路显著下调;流式细胞术进一步证实敲除小鼠肝脏中NK细胞比例减少,且其分泌的IFN-γ、TNF-α和颗粒酶B水平降低;敲除和耗竭试验表明,NK细胞功能丧失可消除AKR1D1敲除与野生小鼠的肿瘤生长差异,明确了NK细胞活性是AKR1D1抑制HCC进展的核心。
3. 接下来对AKR1D1敲除小鼠的肝脏和血清进行代谢组检测,观察到iso-LCA水平显著升高。机制研究发现,iso-LCA通过抑制CREB1磷酸化(p-CREB1)下调细胞因子表达,而过表达iso-LCA可加速野生小鼠肿瘤生长,并降低NK细胞活性。
4. 宏基因组分析发现,AKR1D1敲除小鼠的肠道菌群中B.ovatus丰度显著增加,且与血清iso-LCA水平呈正相关。体外厌氧共培养实验证实,B.ovatus可将初级胆汁酸CDCA转化为iso-LCA。临床样本分析显示,HCC患者粪便中B.ovatus丰度和血清iso-LCA水平升高,且与AKR1D1表达呈负相关。抗生素清除菌群或补充B.ovatus可分别逆转或加剧AKR1D1敲除小鼠的肿瘤进展,证实菌群-胆汁酸轴的核心调控作用。
5. 最后利用结构相似性进行药物筛选,发现螺内酯可竞争性结合胆汁酸受体GPBAR1,逆转iso-LCA对NK细胞的抑制作用,服用此药可作为一种有潜力的HCC辅助治疗方案。
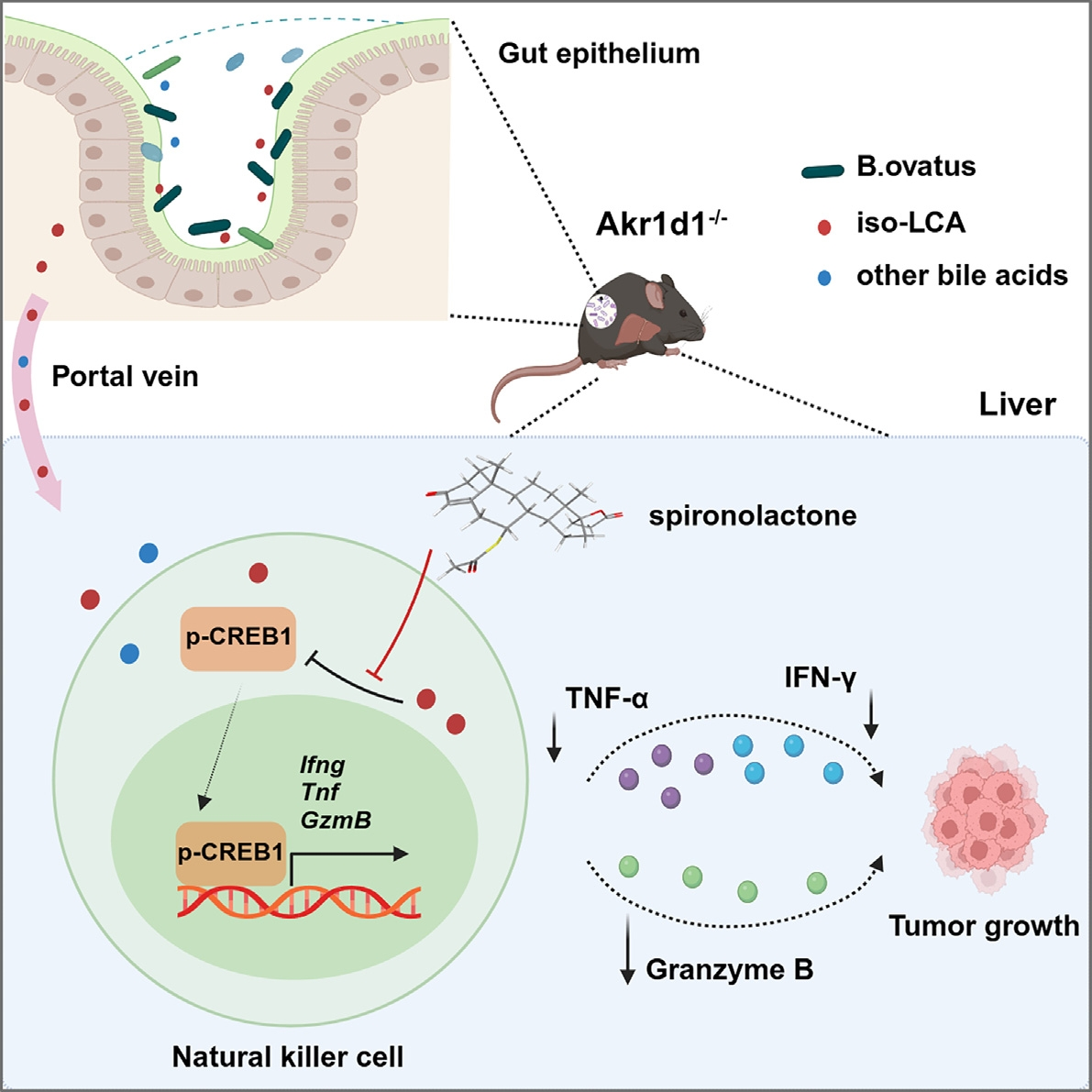
参考文献:Wei H, Suo C, Gu X, et al. AKR1D1 suppresses liver cancer progression by promoting bile acid metabolism-mediated NK cell cytotoxicity. Cell Metabolism. 2025
【3】
Communications Medicine | 二甲双胍调节从血液循环到肠腔的葡萄糖通量

二甲双胍是治疗糖尿病的常用药,但其作用机制尚未完全明确。近年来,研究发现二甲双胍在肠道中的有较高的局部浓度和较长的作用时间,提示其可能通过肠道机制发挥疗效。本研究通过对现有FDG PET-MRI图像的回顾性分析,发现二甲双胍可增加肠道腔内FDG(氟代脱氧葡萄糖)的积累,表明其能刺激葡萄糖向肠道排泄。为此研究人员开展一系列实验,揭示了二甲双胍对Ⅱ型糖尿病患者体内葡萄糖通量的影响,并探讨其潜在机制。
1. 新的葡萄糖通路和通量:对服用或未服用二甲双胍的Ⅱ型糖尿病患者进行PET-MRI检查。连续成像显示,FDG最初出现在空肠,随后向结肠移动。定量分析表明,服用二甲双胍的患者肠道腔内放射性明显增加,且计算得出其葡萄糖排泄率(GER)约为 1.65 g h-1,未服用者约为 0.41 g h-1。给小鼠注射18F标记FDG后,也观察到类似的结果。
2. 肠道微生物代谢葡萄糖:给小鼠注射13C标记的葡萄糖,用GC-MS分析粪便中的短链脂肪酸(SCFAs)。结果表明,肠道微生物能够将肠道中的葡萄糖代谢为短链脂肪酸(SCFAs),且二甲双胍增强了这一过程,这可能有助于肠道微生物与宿主之间的共生关系。
3. 二甲双胍的作用机制:二甲双胍可能通过调节肠道中的葡萄糖通量来发挥作用,这一机制可能与肠道微生物的代谢活动密切相关,为二甲双胍的降糖效果提供了新的解释。
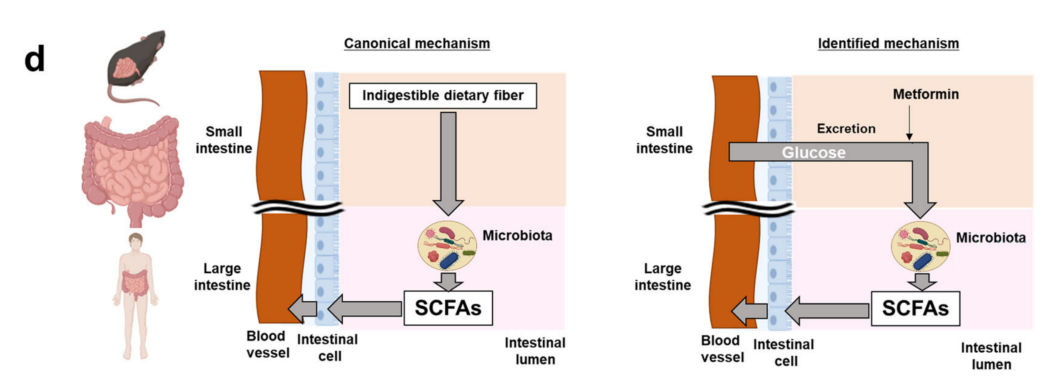
参考文献:Sakaguchi, K., Sugawara, K., Hosokawa, Y., et al. Metformin-regulated glucose flux from the circulation to the intestinal lumen. Communications Medicine. 2025
【4】
Brain Behavior and Immunity | 二甲双胍通过肠道微生物衍生的胆酸代谢物重编程色氨酸代谢,以改善小鼠的抑郁样行为

二甲双胍(Metformin)是一种常用的抗糖尿病药物,也被用作抑郁症的辅助治疗,但其作用机制尚不明确。本研究发现二甲双胍可以通过微生物群-大脑相互作用重编程5-羟色胺代谢以减轻抑郁综合征,提示肠道微生物群衍生的胆汁酸可能成为抑郁症治疗的候选药物。
1. 通过对小鼠的海马组织进行转录组分析,结果发现:CRS(慢性束缚应激组)小鼠的DEGs主要富集在色氨酸代谢和血清素能突触相关通路。二甲双胍激活了抑郁小鼠模型中的MAPK信号、cAMP信号和几种病毒感染途径,且富集到的生物过程与肠道微生物群衍生因子(特别是色氨酸及其代谢产物)高度相关。
2. 通过微生物组分析发现,二甲双胍显著增加CRS小鼠肠道中Akkermansia muciniphila(A. muciniphila)的丰度,在功能层面,二甲双胍治疗后色氨酸代谢得到有效恢复。
3. 将二甲双胍治疗的小鼠的粪便微生物群移植到CRS小鼠中,发现可以改善抑郁样行为,表明肠道微生物在其中发挥关键作用。进一步使用A. muciniphila进行特定微生物干预,发现A. muciniphila移植对受体小鼠的肠道屏障功能障碍和炎症标志物有类似二甲双胍的作用,且增加了海马体中色氨酸和5-HT水平。
4. 通过代谢组学分析发现A. muciniphila能够产生牛磺胆酸和脱氧胆酸代谢产物,这些代谢物在二甲双胍治疗的CRS小鼠中显著增加,且单独给予上述代谢物也可改善CRS诱导的5-HT代谢障碍和抑郁样行为。
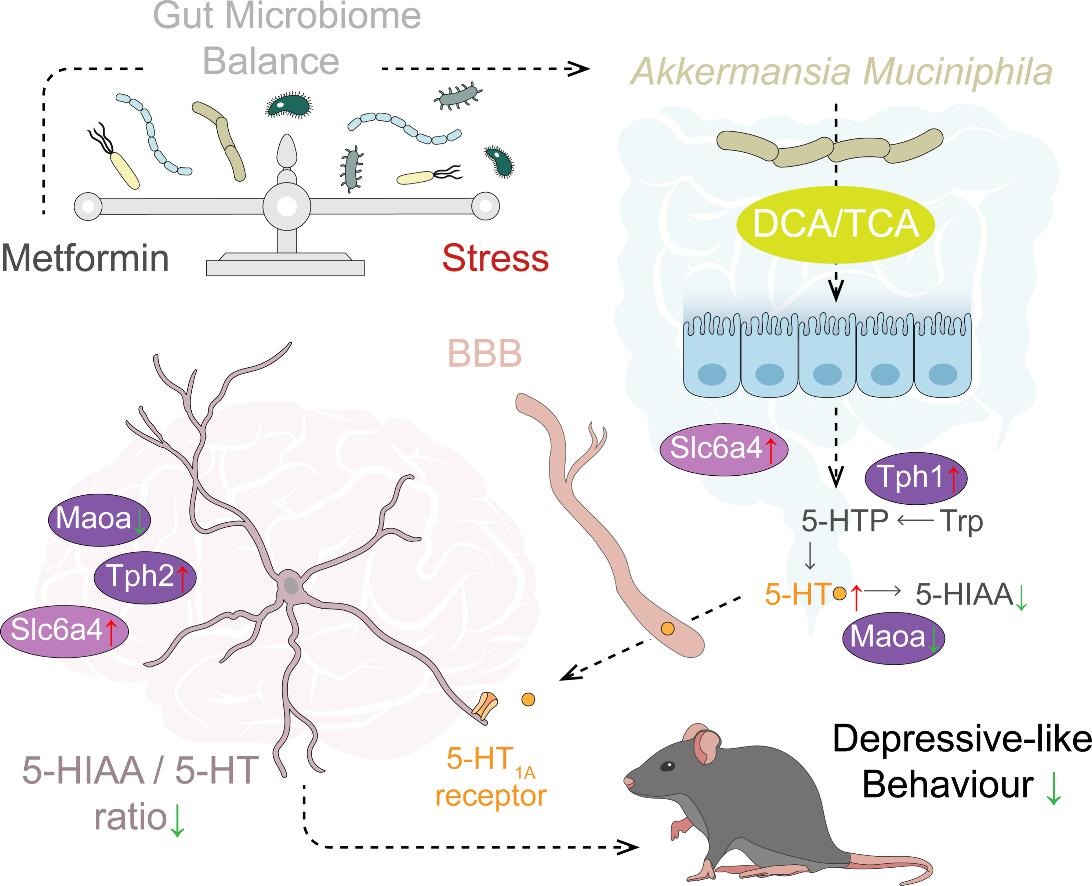
参考文献:Xie X, Li W, Xiong Z, et al. Metformin reprograms tryptophan metabolism via gut microbiome-derived bile acid metabolites to ameliorate depression-Like behaviors in mice. Brain Behavior Immunity. 2025
【5】
Science | 神经酰胺通过FPR2受体的代谢信号传导抑制脂肪细胞产热

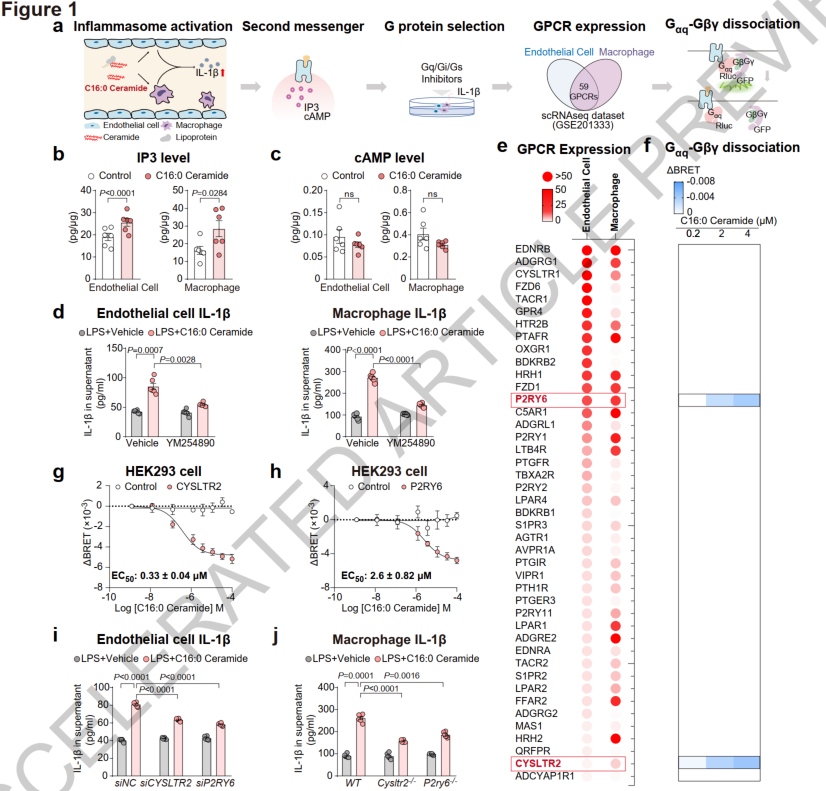
神经酰胺是细胞膜中的重要成分,参与多种细胞过程,且与2型糖尿病、肥胖、脂肪肝、动脉粥样硬化等代谢性疾病密切相关。既往高脂动物实验发现神经酰胺水平在脂肪组织中显著升高,与非酒精性脂肪肝病和糖尿病视网膜病变显著相关。然而,神经酰胺作为细胞间信号分子的机制尚不清楚。本研究探讨了神经酰胺是否通过与G蛋白偶联受体(GPCRs)直接相互作用来调节脂肪细胞的产热功能,特别是以C16:0神经酰胺为例,揭示其在棕色和米色脂肪细胞中的作用机制。
1. FPR2鉴定为神经酰胺的受体:FPR2能够特异性结合长链神经酰胺(C14-C20),并通过Gi-cAMP信号通路抑制脂肪细胞产热。
2. 神经酰胺对产热的抑制作用依赖于FPR2:通过在野生型(WT)小鼠中注射C16:0神经酰胺,研究人员观察到小鼠在寒冷条件下的氧气消耗和能量消耗降低,表明产热功能受损。而在FPR2基因敲除(Fpr2fl/fl)的小鼠中,该效应消失,证实了FPR2在神经酰胺抑制产热中的关键作用。
3. FPR2与神经酰胺的直接相互作用:利用荧光素异硫氰酸酯(FITC)标记的FPR2激动剂进行竞争性结合实验,研究人员证实了C16:0神经酰胺能够直接与FPR2结合,并且这种结合具有较高的亲和力。
4. FPR2的结构基础:研究人员通过单颗粒冷冻电镜技术解析了FPR2与C16:0、C18:0和C20:0神经酰胺形成的复合物结构,这些结构显示,神经酰胺的脂肪酸链深埋在FPR2的正构配体口袋中,并且该口袋具有一定的柔性,能够适应不同长度的神经酰胺。
5. 神经酰胺对FPR2的激活机制:通过比较apo-FPR2模型和神经酰胺-FPR2复合物的结构,研究人员提出了FPR2被神经酰胺激活的可能机制:神经酰胺的结合导致FPR2的跨膜螺旋6(TM6)向外移动,进而影响到G蛋白的激活。

参考文献:Lin, H., Ma, C., Cai, K., et al. Metabolic signaling of ceramides through the FPR2 receptor inhibits adipocyte thermogenesis. Science. 2025
【6】
Nature | 内源性受体CYSLTR2和P2RY6感知神经酰胺来加重动脉粥样硬化

动脉粥样硬化是心血管疾病的主要病因,现有降血脂疗法无法完全消除心血管风险。近年来,研究发现循环中的长链神经酰胺被视为动脉粥样硬化性心血管疾病(ASCVD)的独立风险因素,但它加重ASCVD的机制尚不明确。本研究旨在探究循环长链神经酰胺是否通过激活膜G蛋白偶联受体(GPCRs)来加剧动脉粥样硬化。
1. 受体鉴定:通过构建小鼠动脉粥样硬化模型,结合G蛋白信号量化、GPCRs表达的生物信息分析和NLRP3炎性小体激活的功能检测,发现CYSLTR2和P2RY6是C16:0神经酰胺激活炎性小体的潜在内源性受体,并通过基因敲除、RNA干扰和拮抗剂处理等得到验证。
2. 体内验证:利用FlAsH-BRET技术检测受体的细胞外构象变化,证实神经酰胺能在体内直接结合并激活受体CYSLTR2和P2RY6。
3. 代谢组学分析:慢性肾病(CKD)患者血浆神经酰胺水平升高,且与冠心病严重程度正相关。在小鼠模型中,抑制CYSLTR2和P2RY6可减轻CKD加重的动脉粥样硬化,且不影响胆固醇或神经酰胺水平。
4. 结构解析:运用冷冻电镜技术解析了神经酰胺-CYSLTR2-Gq复合物的结构,揭示了神经酰胺与CYSLTR2的特异性结合模式及其激活机制。
参考文献:Zhang S, et al. Sensing ceramides by CYSLTR2 and P2RY6 to aggravate atherosclerosis. Nature. 2025
【7】
Science Advances | 瓜氨酸调节小鼠巨噬细胞代谢和炎症以对抗衰老

在人口老龄化加剧的当下,衰老相关慢性疾病对健康的威胁日益凸显。代谢失调与衰老紧密相连,然而内源性代谢物在衰老进程中的作用仍有待深入挖掘。本研究旨在探究内源性代谢物与衰老的关系,寻找对抗衰老的潜在干预手段。
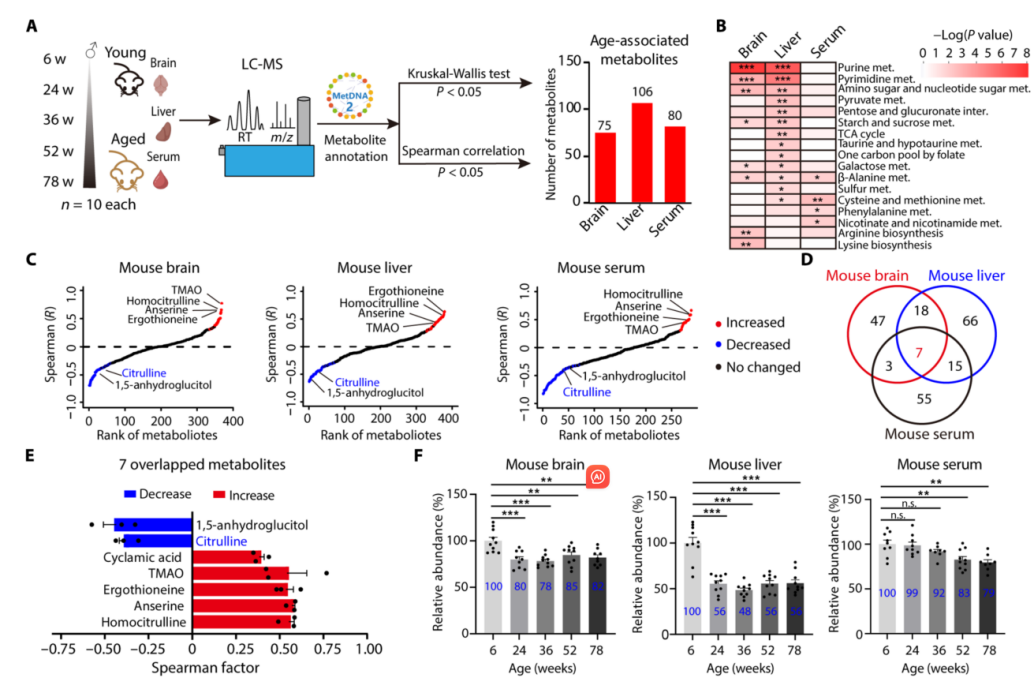
1. 代谢物筛选:采用基于液相色谱-质谱(LC-MS)的非靶向代谢组学技术,分析不同年龄雄性小鼠脑、肝及血清中的代谢物。经Kruskal-Wallis检验和Spearman相关性分析,确定7种与衰老相关的代谢物,其中随着年龄的增长,瓜氨酸的含量会显著下降。
2. 体外实验:在活性氧诱导的细胞衰老模型中,用瓜氨酸处理衰老的小鼠胚胎成纤维细胞(MEF)和原代骨髓来源巨噬细胞(BMDMs)。结果显示,瓜氨酸能减少细胞DNA损伤(γH2AX水平降低),抑制衰老相关β-半乳糖苷酶活性,下调衰老标记物(如p21)和炎症细胞因子(Tnf、Il6、Il1b)的mRNA水平,改善衰老表型。
3. 体内实验:给年轻和老年小鼠长期饮用含瓜氨酸的水,发现老年小鼠体重、脾脏和肝脏重量下降,脑、肝和BMDMs中炎症细胞因子mRNA水平降低,小胶质细胞激活受到抑制,DNA损伤减轻,而年轻小鼠无明显变化,表明瓜氨酸对老年小鼠的衰老相关表型有改善作用。
4. 机制探究:利用代谢组学和蛋白质组学等技术,发现瓜氨酸可调节巨噬细胞代谢,抑制mTOR激活,调控mTOR-缺氧诱导因子1α-糖酵解信号通路,进而对抗衰老和炎症。采用稳定同位素示踪代谢组学技术,以[U-13C]精氨酸作为示踪剂,发现衰老过程中巨噬细胞瓜氨酸缺乏与Nos2表达下降有关。
参考文献:Xie Z, et al. Citrulline regulates macrophage metabolism and inflammation to counter aging in mice. Science Advances. 2025
【8】
Cell Metabolism | 麦角硫因通过激活MPST调控线粒体功能和运动表现

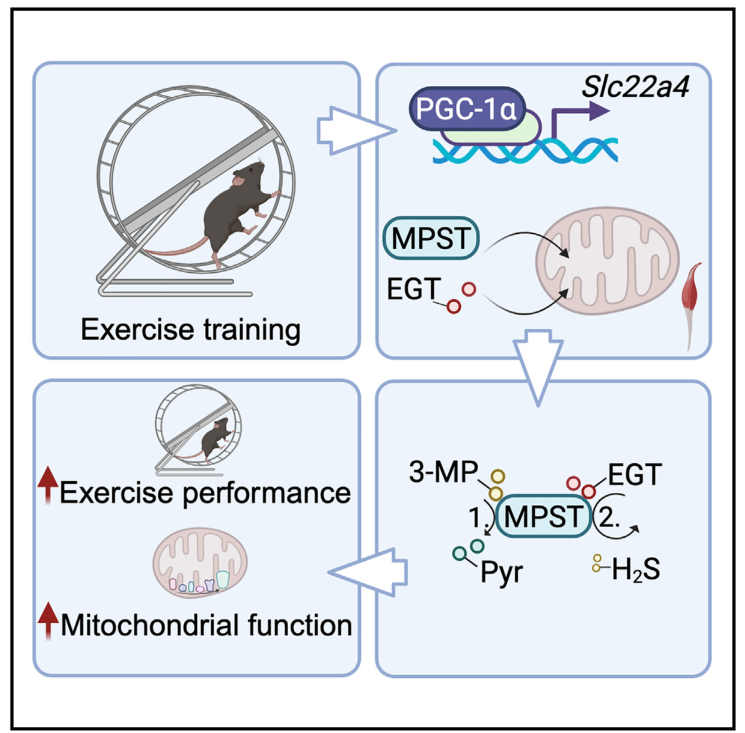
麦角硫因(EGT)是一种来源于饮食的非典型氨基酸,广泛存在于人体组织中。研究表明,EGT水平降低与多种年龄相关疾病(如神经退行性疾病和心血管疾病)有关,而EGT补充剂则表现出保护作用。然而,EGT的直接作用靶点一直不清楚。本研究通过代谢组和蛋白组研究确定了3-巯基丙酮酸硫转移酶(MPST)是EGT的直接分子靶点,为理解运动训练的分子机制和开发新的运动增强策略提供了新的见解。
1. 通过对为期4周训练后的MITO-Tag小鼠进行代谢组学分析,在肌肉组织线粒体中检测到81种代谢物,包括NAD、NADH、NADP、NADPH和α-酮戊二酸。
靶向代谢组学进一步确认EGT在运动训练肌肉线粒体中最显著富集,与久坐肌肉组织线粒体相比几乎增加了两倍。
2. 采用PISA技术识别蛋白质-配体相互作用,发现最稳定的两种蛋白质都是线粒体呼吸调节因子——MPST和CYCS(细胞色素C),且MPST结合程度更高。随后利用纯化的人重组MPST蛋白,通过核磁共振(NMR)实验,进一步验证了EGT与MPST之间的直接结合。
3. 通过给运动训练期间的小鼠喂食富含EGT的饲料,结果显示,EGT补充剂可以显著增加小鼠的耐力运动表现,包括增加运动速度和总运动距离,并且对肌肉代谢产生了显著影响。
参考文献:Sprenger HG, Mittenbühler MJ, Sun Y, et al. Ergothioneine controls mitochondrial function and exercise performance via direct activation of MPST. Cell Metabolism. 2025