PICRUSt(phylogeneticinvestigation of communities by reconstruction of unobserved states)通过隐性状态重建进行群落系统进化的研究,该软件基于16S rDNA和参考序列数据库,预测宏基因组功能组成。
PICRUSt的原理基于已测细菌基因组的16S rRNA全长序列,推断它们的共同祖先的基因功能谱,对Greengenes数据库中其它未测物种的基因功能谱进行推断,构建古菌和细菌域全谱系的基因功能预测谱;最后,将测序得到的菌群组成“映射”到数据库中,对菌群代谢功能进行预测(详见下图)。
为了能够通过16S测序数据来准确的预测出功能构成,首先需要对原始16S测序数据的种属数量进行标准化,因为不同的种属菌包含的16S拷贝数不相同。然后将16S的种属构成信息通过构建好的已测序基因组的种属功能基因构成映射获得预测的功能结果。
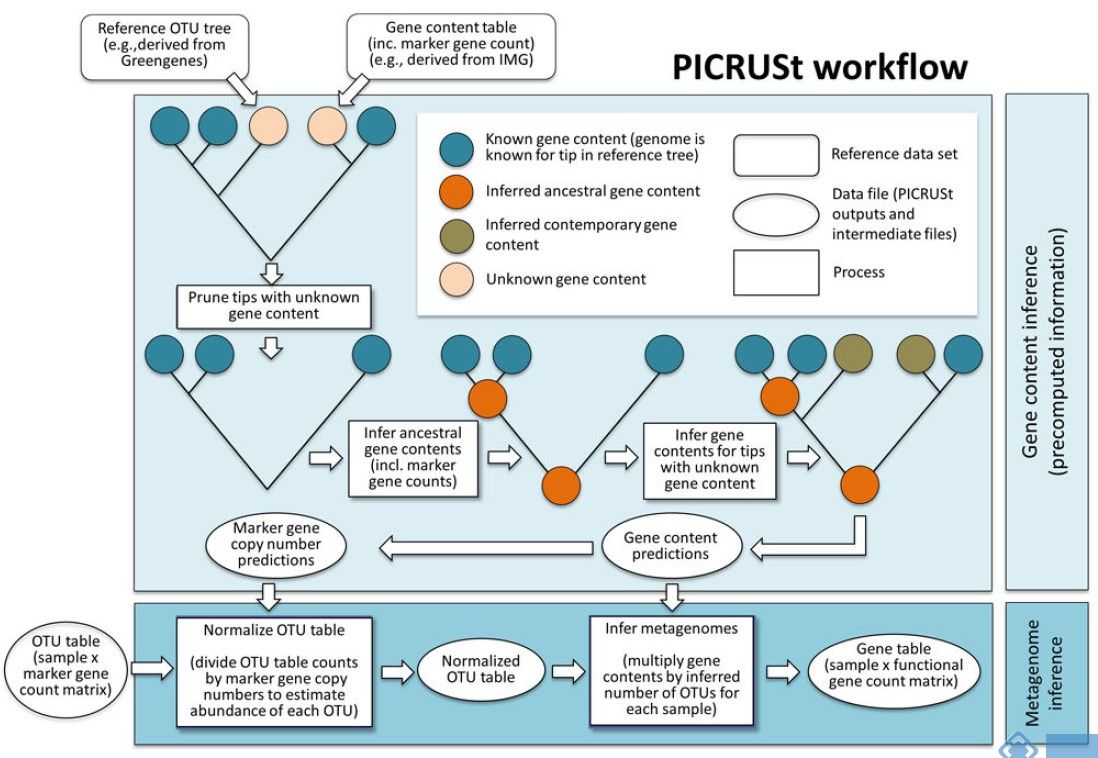
图 PICRUSt工作流程
该分析的缺点是古菌和细菌域全谱系的基因功能预测谱是基于Greengenes数据库进行构建的,Greengenes版本为gg_13_5,已经长时间未更新,因此很多古菌和细菌并未包含在内。此外,该预测结果只能预测到KEGG某个pathway水平,但不能从基因层面预测研究。如果在做宏基因组研究之前,想先看下关注的pathway是否存在或是否在不同分组中存在显著性差异,此时可以先做下功能预测,基于功能预测结果进行后续宏基因组实验设计。
PICRUSt在线分析网址:
http://huttenhower.sph.harvard.edu/galaxy/
具体使用步骤如下:
1.输入数据格式
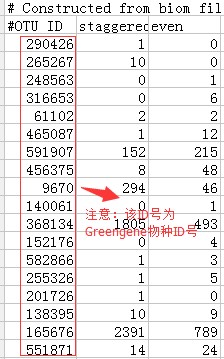
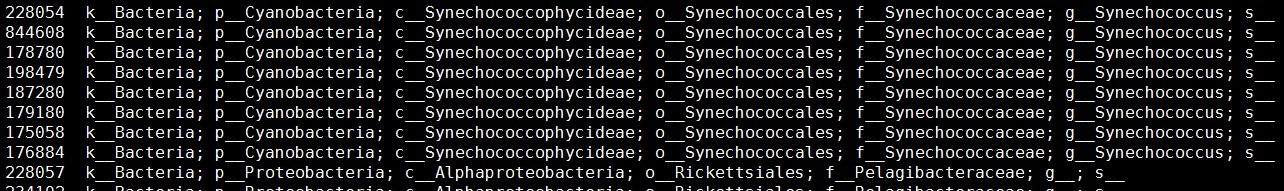
注:数据输入格式第一列为Greengene物种ID号,第二列-第n列为不同样本的表达量。如果OTU注释所用的数据库不是Greengene,需要根据OTU物种注释信息,转换为GreengeneID。Greengene对应的物种信息下载网址如下:
http://greengenes.secondgenome.com/downloads/database/13_5
(由于Greengene长时间未更新,用其他数据库注释得到的物种可能在Greengene物种列表中不存在)
2.将数据上传至网页(需要注意的是数据类型需要选择:picrust)
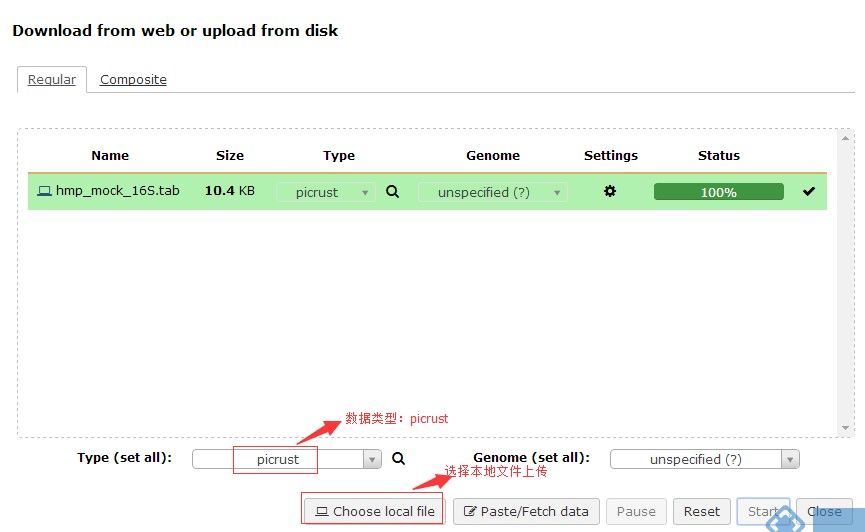
3.Normalize By Copy Number
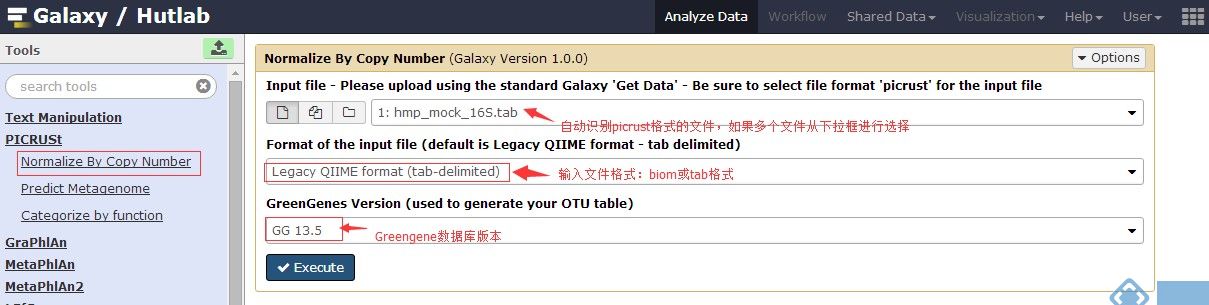
4.Predict Metagenome(在预测分类中,按需选择 “KEGGOrthologs”、“COG”或者“Rfam”)
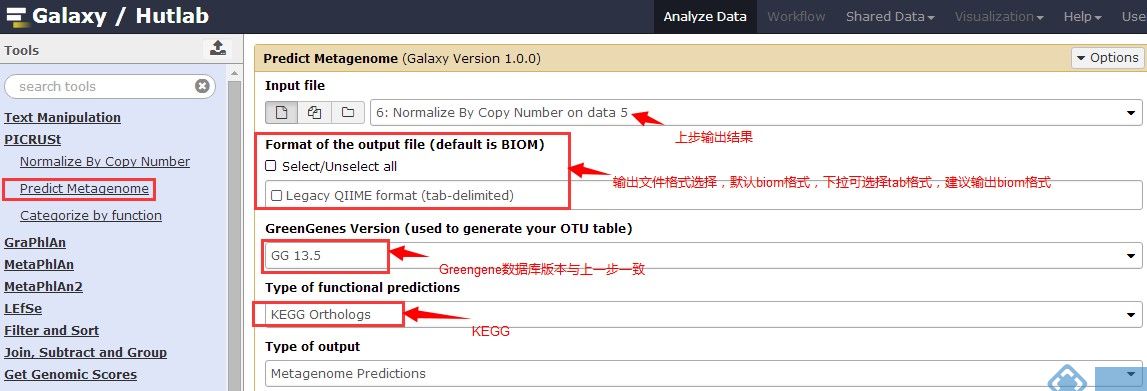
5.Categorize by function
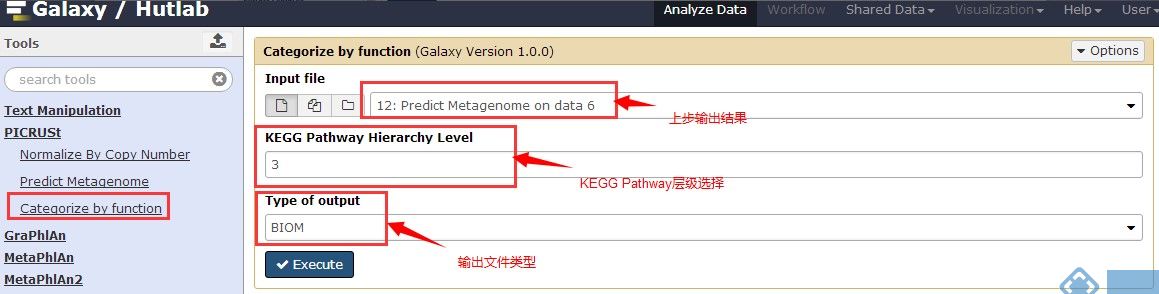
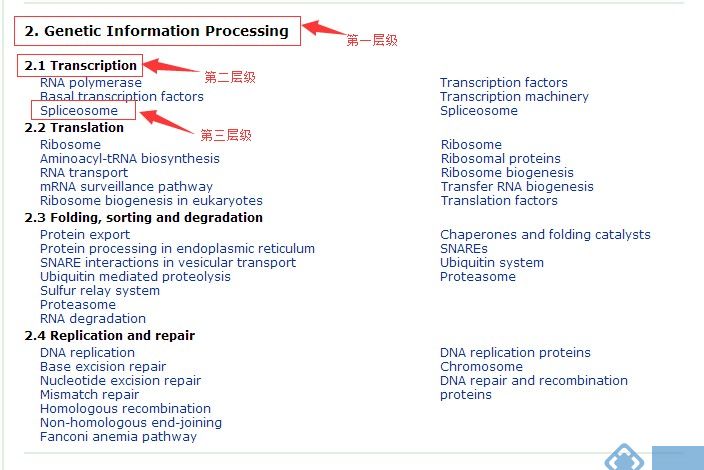
其中“KEGG Pathway HierarchyLevel“有三种层级(具体的层级含义见下图)可选,输出文件格式建议使用更常见的格式“Legacy QIIME format(tab)”。
KEGG Pathway网址:http://www.kegg.jp/kegg/pathway.html
6.基于预测结果,统计不同分组间显著性差异的KEGG Pathway(该分析可直接使用Windows本地软件STAMP(下文会对该软件进行详细使用说明)实现)
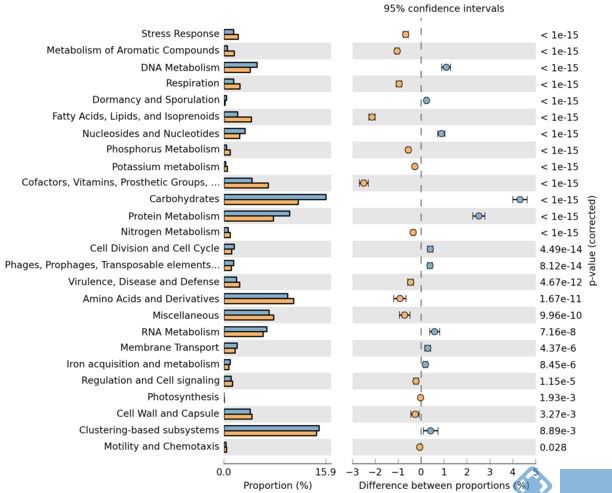
至此,16S在线功能预测分析就完成了,有没有觉得很简单呢?如果16S功能预测的结果不能满足您对微生物功能层面的研究,建议选择合适的样本进行宏基因组或宏转录组测序分析,会获得意想不到的收获。
相关阅读:
